
This is the first in a series of five articles written jointly by Pharmagro and Terriva. We focus on the importance of closing the gap between assumed capacity and actual performance and on the need for process robustness.
In their report CDMO Growth Report 2025, the consultancy Simon Kucher assessed the elements that make for successful collaboration between pharma companies and CDMOs. The graph shows how each side rated ten criteria. Interestingly the CDMOs overestimated the need for speed (ability to meet tight deadlines) while underestimating supply chain reliability. Both sides were in close agreement on the need for smooth scale up.
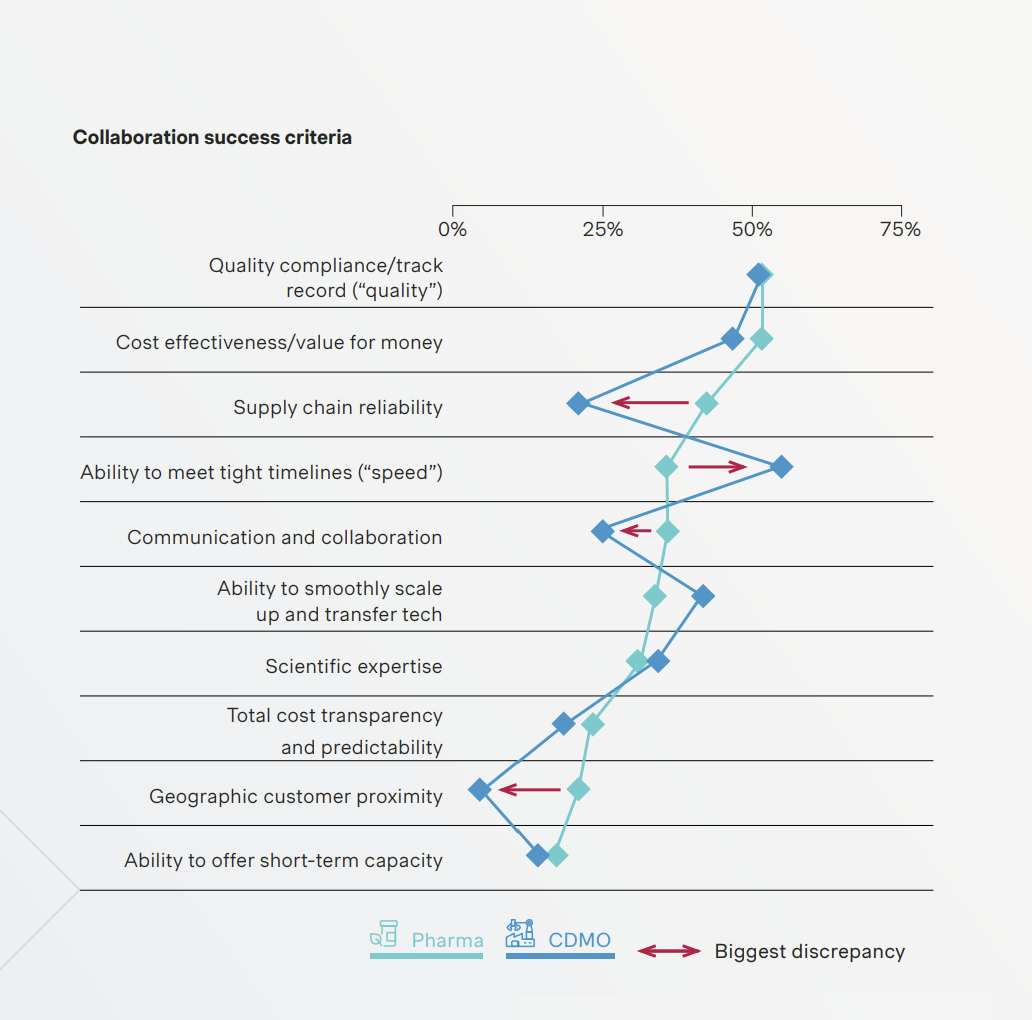
Source : CDMO Growth Report 2025 Simon Kucher
Having sat in board meetings and seen the consequences of failed PPQs or late tech transfers, we will cover broad topics of the trade off between speed and reliability, and will deep dive into examples of equipment choice.
The five articles will cover
- Audit Ready By Design – What CDMOs miss about CFR21 in Equipment Selection
- Tech Transfer and Process Inconsistency – The Example of Blending
- Speed – Not At Any Cost
- Containment & Control
- Scaling from R&D to Commercial
Terriva provides state of the art powder blending equipment and control systems. Pharmagro provides software and services for business development and portfolio management. Both companies share a data driven approach to scaling up, whether it is equipment or processes.
Article 1 – Audit Ready By DEsign – What CDMOs miss about 21 CFR compliance in equipment selection
There are considerable capacity expansions being announced by CDMOs, particularly in the areas of high spec small molecule manufacturing and sterile fill / finish. Installation of the equipment is one thing, but as important are the software, process controls and training of the operators.
The Illusion of Compliance
Most Contract Development and Manufacturing Organisations (CDMOs) believe they are 21 CFR Part 11 compliant.
Until an audit proves otherwise.
The assumption is simple:
If the software is compliant, the system is compliant.
But auditors don’t assess software in isolation.
They assess system behaviour, data integrity, and how equipment performs in real-world conditions.
That’s where the gap exists.
And it’s why compliance failures are often discovered not during validation — but under audit pressure.
The Core Misconception: “Compliance is a Software Layer”
21 CFR Part 11 is frequently treated as a feature set:
- Electronic signatures
- User access controls
- Data storage
But the reality is more demanding:
“Can your system prove, without ambiguity, what happened, when it happened, and who was responsible?
That is not just software.
That is how the system is designed, integrated, and controlled.
If the underlying equipment and control architecture don’t support this — compliance is fragile at best.
What Auditors Actually Test
Audits are not theoretical exercises.
They are designed to expose weaknesses in real-world operation.
Auditors will typically focus on:
Data Integrity (ALCOA+)
“Data integrity ensures that data remains accurate, complete, and reliable throughout its lifecycle. In pharmaceutical manufacturing, this is supported by the ALCOA+ principles, which require data to be attributable, legible, recorded at the time of activity, original, and accurate—ensuring it can be trusted for audits and decision-making.
- Is data attributable, legible, contemporaneous, original, and accurate?
- Can data be altered without trace?
Audit TRails
- Are all critical process parameters automatically recorded?
- Are changes logged in real time?
- Can records be edited, overwritten, or deleted?
User Permissions & Access Control
- Are roles clearly defined and enforced?
- Is there segregation of duties?
- Are actions traceable to individuals?
System Behaviour Under Operation
- Does the system enforce process parameters?
- Can operators override steps without traceability?
- Are deviations captured and justified?
Data Retention & Retrieval
- Can batch records be retrieved quickly and securely?
- Is data protected, backed up, and exportable?
The key point:
Auditors are not checking if a system can be compliant —
they are testing whether it is compliant in practice.
Where CDMOs Get Caught Out
Most compliance gaps don’t come from intent.
They come from equipment decisions made without fully understanding downstream risk.
Common issues include:
- Incomplete audit trails (manual inputs, non-continuous data capture)
- Weak access control (shared logins, untracked overrides)
- Disconnected systems (manual data transfer, multiple data sources)
- Unenforced processes (operators able to adjust parameters without trace)
These gaps are rarely visible at purchase stage.
But they become highly visible during an audit.
The Real Issue: Compliance Risk is Introduced at Selection
In CDMO environments, equipment decisions are rarely isolated.
They must support:
- Multiple customers
- Multiple product types
- Varying regulatory expectations
- Long lifecycle requirements
Which means:
“Every equipment decision carries portfolio-level risk.

Pharmagro Perspective: Compliance Risk Starts at the Investment Decision
From Pharmagro’s perspective, 21 CFR Part 11 is not just a validation topic.
It is a strategic risk consideration embedded within portfolio and investment decisions.
Before equipment is selected, CDMOs should be asking:
- Is this system aligned with the molecules and processes we intend to support?
- Does it introduce hidden compliance or scale-up risk?
- Can it deliver consistent, repeatable performance across multiple programmes?
How will this impact timelines, regulatory exposure, and commercial outcomes?
Because in practice:
“Compliance failures are often a symptom of earlier investment decisions.
Through technical due diligence and portfolio-level thinking, Pharmagro help CDMOs identify where:
- System design may compromise data integrity
- Control limitations may introduce future audit exposure
- Equipment-process misalignment may lead to inconsistency at scale
In a CDMO model, where flexibility and speed are critical, these risks compound quickly.
Terriva Perspective: Engineering Compliance Into the System
Once the right investment decision is made, compliance must be engineered into the equipment itself.
From Terriva’s standpoint, 21 CFR Part 11 readiness is not an add-on — it is built into:
Control System Architecture
- Secure, tamper-evident audit trails
- Role-based user access
- Electronic records and signatures aligned to regulatory expectations
Process Enforcement
- Defined recipes with locked parameters
- Controlled operator interaction
- Full traceability of any deviation
Data Integrity by Design
- Automated, continuous data capture
- Secure storage and structured export
- Integration-ready systems for wider digital environments
This ensures that the system doesn’t just perform — it can prove compliance under scrutiny.
Key Insight: Compliance Isn’t Added Later — It’s Engineered In
This is the shift CDMOs need to make.
21 CFR Part 11 is not:
- A software upgrade
- A validation exercise
- A documentation process
It is a system-level design principle.
And the earlier it is addressed, the lower the risk. Furthermore, the use of Artificial Intelligence in manufacturing requires a base of reliable data that can be kept up to date. Therefore, 21 CFR compliance is an important contributor to this.
Practical Questions CDMOs Should Be Asking
Before selecting equipment, ask:
- Can this system demonstrate data integrity under audit conditions?
- Are audit trails automatic, secure, and tamper-evident?
- Is user access fully controlled and traceable?
- Does the system enforce process discipline, not rely on operator behaviour?
- Will this system support multiple products and regulatory expectations over time?
If any of these answers are unclear —
you are introducing risk into your operation.
Conclusion: Designing for Audit, Not Just Operation
In today’s CDMO landscape:
- Regulatory scrutiny is increasing
- Customer expectations are rising
- Data integrity is non-negotiable
The difference between passing and failing an audit is rarely effort.
It is design.
“Audit-ready systems are not built during validation.
They are defined at the point of investment — and engineered at the point of delivery.
Joint Positioning
- Pharmagro support CDMOs in making the right investment decisions by identifying portfolio, technical, and compliance risk upfront
- Terriva ensures those decisions are realised through equipment engineered for audit-ready performance
Pharmagro define the risk. Terriva engineer it out.
If you are reviewing your equipment strategy or planning future investment:
Speak to Pharmagro and Terriva about building systems that don’t just perform — but stand up to audit.
FAQs: 21 CFR Part 11 & Equipment Selection for CDMOs
Is 21 CFR Part 11 compliance just about software?
No. 21 CFR Part 11 is not just a software requirement. While software enables features such as electronic signatures and audit trails, compliance is ultimately about system behaviour, data integrity, and how equipment operates in practice. If the underlying equipment and control system are not designed to enforce compliant processes, software alone cannot ensure compliance.
What do auditors actually look for during a 21 CFR Part 11 inspection?
Auditors focus on how systems behave under real operating conditions. Key areas include data integrity based on ALCOA+ principles, secure and tamper-evident audit trails, role-based user permissions, traceability of operator actions, and secure data storage and retrieval. They are not just checking features, they are testing whether the system can prove compliance consistently.
Why is equipment selection important for 21 CFR Part 11 compliance?
Equipment selection is critical because compliance risk is often introduced at the point of investment. If a system lacks the right control architecture, audit trail capability, or process enforcement, those gaps become difficult and costly to fix later. Choosing the right equipment helps ensure compliance is built in from the start.
What are the most common compliance gaps in blending systems?
Common compliance gaps include:
- Incomplete or manual audit trails
- Shared user logins or weak access control
- Lack of automated data capture
- The ability for operators to change parameters without traceability
- Disconnected systems requiring manual data handling
These issues may not be obvious initially but can lead to significant audit findings.
Can 21 CFR Part 11 compliance be added after installation?
It can be attempted, but it is rarely the most efficient or effective approach. Retrofitting compliance often requires control system upgrades, additional validation work, and increased cost and downtime. The most reliable approach is to ensure compliance is engineered into the system from the beginning.
How do CDMOs reduce compliance risk when investing in new equipment?
CDMOs can reduce compliance risk by:
- Defining clear User Requirement Specifications
- Assessing equipment against data integrity and audit requirements
- Carrying out technical due diligence on system capabilities
- Ensuring alignment with current and future product portfolios
This helps reduce the risk of selecting equipment that cannot support long-term compliance.
How does system design impact data integrity?
System design determines how data is captured, stored, protected, and retrieved. Poorly designed systems may rely on manual input, lack secure audit trails, or allow untracked changes. Well-designed systems support automated data capture, tamper-evident records, and full traceability of actions, which are essential for meeting regulatory expectations.
What role does process enforcement play in compliance?
Process enforcement ensures that operators follow defined procedures without unauthorised deviation. A compliant system should lock critical parameters, prevent unauthorised changes, and record any deviations with justification. Without process enforcement, compliance depends too heavily on operator behaviour, which creates risk.
Why is 21 CFR Part 11 particularly challenging for CDMOs?
21 CFR Part 11 can be particularly challenging for CDMOs because they operate in complex environments with multiple clients, diverse product types, and varying regulatory expectations. This increases the need for systems that are flexible, scalable, and consistently compliant across multiple programmes.
What is the key takeaway for CDMOs when selecting equipment?
The key takeaway is that compliance is not something to add later. It should be engineered into the system from the outset. CDMOs should prioritise equipment that not only performs operationally but can also demonstrate compliance under audit conditions.


